the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Nov 2018
| 08 Nov 2018
A 3-year time series of volatile organic iodocarbons in Bedford Basin, Nova Scotia: a northwestern Atlantic fjord
Qiang Shi
Douglas Wallace
We report weekly observations of volatile organic iodocarbons (CH3I, CH2ClI and CH2I2) over the time period May 2015 to December 2017 from four depths in Bedford Basin, a coastal fjord (70 m deep) on the Atlantic coast of Canada. The fjord is subject to wintertime mixing, seasonal stratification and bloom dynamics, subsurface oxygen depletion, local input of freshwater, and occasional intrusions of higher-density water from the adjacent continental shelf. Near-surface concentrations showed strong seasonal and sub-seasonal variability, which is compared with other coastal time series. The vertical variation of CH2I2 and CH2ClI within the upper 10 m is consistent with rapid photolysis of CH2I2. Average annual sea-to-air fluxes (46.7 nmol m−2 day−1) of total volatile organic iodine were similar to those observed in other coastal and shelf time series, and polyiodinated compounds contributed 80 % of the total flux. Fluxes were subject to strong interannual variability (a factor of 2) mainly due to wind speed variability. Near-surface net production of CH3I averaged 1 pmol L−1 day−1 and was similar to rates in the English Channel but an order of magnitude higher than in shallow waters of the Kiel Fjord, Germany, possibly due to higher microbial degradation in the latter. The near-bottom (60 m) time series showed evidence of CH3I production associated with organic matter degradation and a possible “switch” from the production of CH3I via an alkylation pathway to the production of CH2I2 by a haloform-type reaction. Near-bottom CH3I production varied strongly between years but was generally ca. 20 times lower than near-surface production.
- Article
(5384 KB) - Full-text XML
-
Supplement
(145 KB) - BibTeX
- EndNote





Volatile organic iodocarbons (VOIs) such as methyl iodide (CH3I), chloroiodomethane (CH2ClI) and diiodomethane (CH2I2) have a predominantly oceanic source and supply a significant amount of iodine to the atmosphere (see review by Saiz-Lopez and Von Glasow, 2012). These gases, also referred to as VSLSs (very short-lived halogenated substances) due to their reactivity and short atmospheric lifetimes, have been implicated in supporting catalytic ozone destruction in the troposphere (Davis et al., 1996; McFiggans et al., 2000) and potentially in the lower stratosphere (Solomon et al., 1994) as well as aerosol formation in the marine boundary layer (McFiggans et al., 2000, 2004; O'Dowd et al., 2002). Recent modeling of atmospheric reactive iodine (IOx = IO + I) and experimental studies (Carpenter et al., 2013; Jones et al., 2010; Mahajan et al., 2010) suggest that the supply of volatile organoiodine represents <50 % of the total sea-to-air delivery of reactive iodine, with most being supplied in the form of HOI and I2. Nevertheless, the potential for localized higher emissions coupled with their relatively long lifetimes (compared to I2 and HOI) allows the organic compounds to be a significant source of iodine to the free troposphere and even potentially to the lower stratosphere in certain regions (Tegtmeier et al., 2013). Further, Mahajan et al. (2012) noted a strong correlation of IOx and CH3I, suggesting that the sources of CH3I and the shorter-lived precursors of IOx are closely related or depend on similar variables.
CH3I is the most abundant VOI species in the atmosphere (Yokouchi et al., 2011) because of its longer lifetime (days) compared to CH2ClI (h) and CH2I2 (min) (Mössinger et al., 1998; Rattigan et al., 1997). However, the total supply of organically bound iodine to the atmosphere is several times larger than the CH3I supply alone (Carpenter et al., 2014) with the bulk of the remainder transported in the form of CH2I2 and CH2ClI (additional iodocarbons such as CH3CH2I, CH2BrI and CHI3 are generally present in much lower concentrations). Despite considerable attention to the oceanic distribution and sea-to-air flux of these compounds, in particular CH3I (Ziska et al., 2013), it is not yet possible to apportion the oceanic production of these compounds unequivocally to specific mechanisms. Even for CH3I, controversy remains, for example, as to the relative importance of direct “biological” or “photochemical” production pathways with experimental evidence reported for both and correlation analysis generally being inconclusive, in part because of the “snapshot” nature of most studies (Stemmler et al., 2014). Comparisons of models to observed distributions have also proven ambiguous, with localized studies suggesting the predominance of a biological production pathway (Stemmler et al., 2013) but a global analysis emphasizing photochemical production as the dominant mechanism. This diversity of views has been maintained through a variety of experimental studies (Amachi et al., 2001; Brownell et al., 2010; Hughes et al., 2011; Manley and de la Cuesta, 1997; Moore and Tokarczyk, 1993; Moore and Zafiriou, 1994; Richter and Wallace, 2004; Shi et al., 2014a; Smythe-Wright et al., 2006).
For compounds other than CH3I, similar uncertainty exists concerning production pathways, but with fewer underlying studies. Laboratory experiments have shown that the presence of dissolved iodide and dissolved organic matter can lead to the production of these compounds in the dark (Martino et al., 2009). Fuse et al. (2003) and Martino et al. (2005) observed that CH2ClI could be produced by photolysis of CH2I2 in artificial and natural seawater. However, detailed mechanisms and their relative importance in the field remain unclear.
Time series observations can reveal processes and controlling factors underlying the production and loss of iodocarbons in the ocean and provide data for testing hypotheses and/or models. However, very few long-term time series observations of iodocarbons have been reported to date, all from coastal waters. Klick (1992) reported 13 months of weekly measurements of CH2I2 and CH2ClI from very shallow (3.5 m) water in the Kattegat at the Swedish coast. Orlikowska and Schulz-Bull (2009) reported a year of weekly data for CH2ClI, CH2I2, CH3I and C2H5I from a nearshore (3 m of depth) site in the Baltic Sea. Archer et al. (2007) reported a seasonal study of CH2ClI, CH2I2, CH3I, C2H5I and CH2BrI measured weekly at four depths (0–50 m) in the western English Channel from July 2002 to April 2004. Shi et al. (2014b) reported on the seasonal cycle of CH3I from surface waters of the Kiel Fjord: a shallow (14 m), brackish water body in northern Germany, which was sampled weekly for 2 years. Shimizu et al. (2017) presented a time series of vertical profiles (0–90 m) of CH2I2, CH2ClI, CH3I and C2H5I from the center of Funka Bay, Japan, which were measured every 2–4 weeks from March 2012 to December 2014.
Here, we report weekly observations of CH3I, CH2ClI and CH2I2 made over the time period May 2015 to December 2017 at four depths (0–60 m) in Bedford Basin: a coastal fjord on the east coast of Canada. We report seasonal to interannual variability of the observed concentrations at different depths in the water column and compare our results with the other time series. We report daily average fluxes to the atmosphere and use a simple, time-varying mass balance model for near-surface waters to estimate production rates and their variability. We discuss the observed variability of both concentrations and production rates in light of earlier studies, potentially correlated variables and suggested production pathways.
Time series measurements of VOIs were carried out in the Bedford Basin (44.69∘ N, 63.63∘ W) near Halifax, Canada. Bedford Basin is an 8 km long, 17 km2 fjord with a maximum depth of 71 m and a total volume of 500 km3. The Bedford Basin is connected with continental shelf waters of the Atlantic Ocean through “the Narrows” (a ca. 300 m wide and 20 m deep passage; Fig. 1). The basin receives freshwater primarily from the Sackville River at its northwestern end, with a total average freshwater input of 5.41 m3 s−1 (Buckley and Winters, 1992). The average near-surface salinity within the basin is 29, which can be compared with salinities of >30 over the adjacent Scotian Shelf. There are only relatively small horizontal gradients of near-surface salinity within the Bedford Basin itself (typically <2 difference from close to the Sackville River mouth to the Narrows).
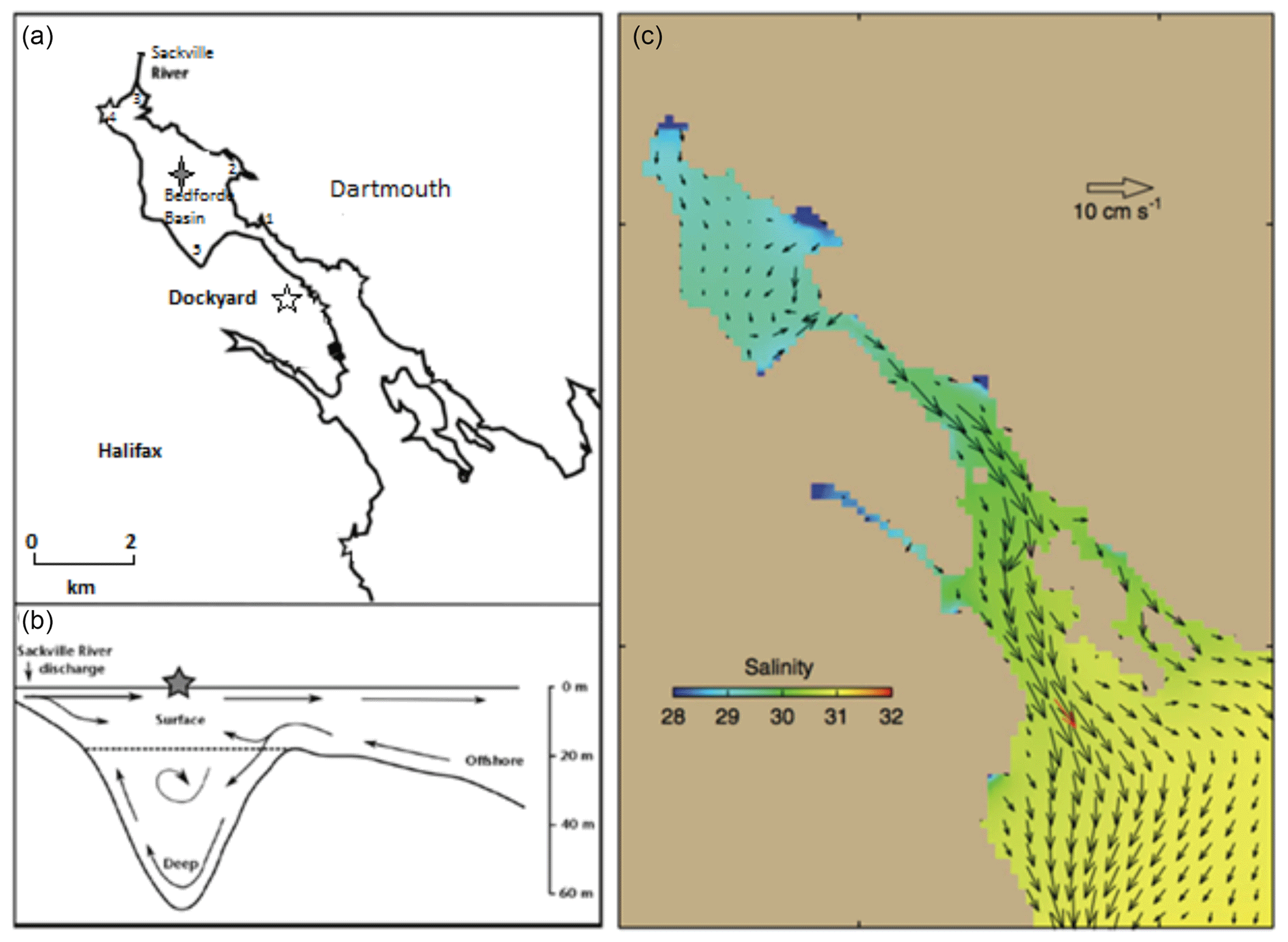
Figure 1(a) Main sampling location (grey star) and nearshore sampling locations 1–5 (1: Tufts Cove; 2: Wrights Cove; 3: Sackville; 4: Mill Cove; 5: Fairview Cove) in Bedford Basin; (b) two-layered flow in Halifax Harbour adapted from Kerrigan et al. (2017); (c) horizontal circulation of water in Halifax Harbour from Shan et al. (2011) using annual mean currents and velocities.
Time series observations of physical, chemical and biological parameters have been recorded since 1992 (Li, 1998). Our halocarbon samples were collected weekly in the center of the Bedford Basin at its deepest point (Fig. 1) between May 2015 and January 2018. Samples were collected with 10 L Niskin bottles attached to a rosette sampler at 1, 5, 10 and 60 m (10 m samples were collected biweekly from May to September 2015). The upper three water samples covered the majority of the euphotic zone. The 60 m water sample was from typically stagnant near-bottom water, which is renewed by vertical mixing events in late winter, and by occasional intrusions of higher-salinity continental shelf water in both summer and winter. Chlorophyll a (Chl a), dissolved oxygen and nutrients were measured weekly at the four depths as part of the Bedford Basin Monitoring Program (details can be found in website: http://www.bio-iob.gc.ca/science/monitoring-monitorage/bbmp-pobb/measurements-mesures-en.php, last access: November 2018). In addition to the Niskin bottle sampling, vertically continuous measurements of temperature, salinity, dissolved oxygen and Chl a properties were measured with a CTD mounted on the rosette. Additional information concerning the measurements of supporting physical and biological parameters can be found in the paper by Burt el al. (2013). Nutrients were measured using a Skalar SAN autoanalyzer; the precisions of , and were ±0.01, ±0.14 and ±0.14 µmol L−1, respectively. Chl a concentration was analyzed using a fluorescence technique (Turner Design model 10 fluorometer) with the root mean square error (RMSE) being 0.23 µg L−1. Dissolved oxygen concentrations were determined at four depths using Winkler titration with a precision of ±0.5 µmol kg−1. The resulting between-lab agreement approaches the specifications for repeat hydrography required by the Global Ocean Observing System (http://www.goosocean.org/components/com_oe/oe.php?task=download&id=35904&version=2.0&lang=1&format=1, last access: November 2018).
The concentrations of iodomethane (CH3I), chloroiodomethane (CH2ClI) and diiodomethane (CH2I2) reported here, as well as of a number of other halocarbons (data not shown), were measured using purge and trap gas chromatography with detection by both mass spectrometry (MS) and electron capture (ECD). All measurements were made using an Agilent Technologies gas chromatograph (GC 7890B) equipped with a capillary column (RTX-VGC; 60 m; 1.4 µm coating, column diameter: 0.25 mm; helium carrier gas 0.5 mL min−1), together with an automated purge and trap system equipped with an autosampler (VSP4000; IMT, Vohenstrauss, Germany). The GC column was temperature programmed as follows: initial temperature 50 ∘C for 6 min, then ramped to 150 ∘C at 6 ∘C min−1 and finally ramped to 200 ∘C at 10 ∘C min−1. Water samples (10 mL) were stored in 20 mL vials equipped with an ultralow-bleed septum prior to purging with helium (20 mL min−1 for 18 min). Every sample was analyzed in triplicate. The standard deviation of triplicate measurements (integrated peak area) was <10 % for CH3I, <15 % for CH2ClI and <20 % for CH2I2.
Calibration of the GC system for CH3I, CH2ClI and CH2I2 was performed using permeation tubes (VICI; Houston, TX, USA), which were maintained at a constant temperature of 23 ∘C and weighed every 2 weeks. Dilutions of the permeation tube effluent were made in ultrahigh-purity N2 (>99.995 %) with flow rates of 50 to 700 mL min−1, and samples were injected into the purge and trap system (VSP) through a 140 µL loop. The standard deviation of the peak area during these calibration runs was <5 % for CH3I and CH2ClI and <15 % for CH2I2. Overall, the calibration response varied by less than 15 % over the entire sampling period.
Throughout the paper, seasons are defined as follows: summer is June through August; fall is September through November; winter is December through February; and spring is March to May.
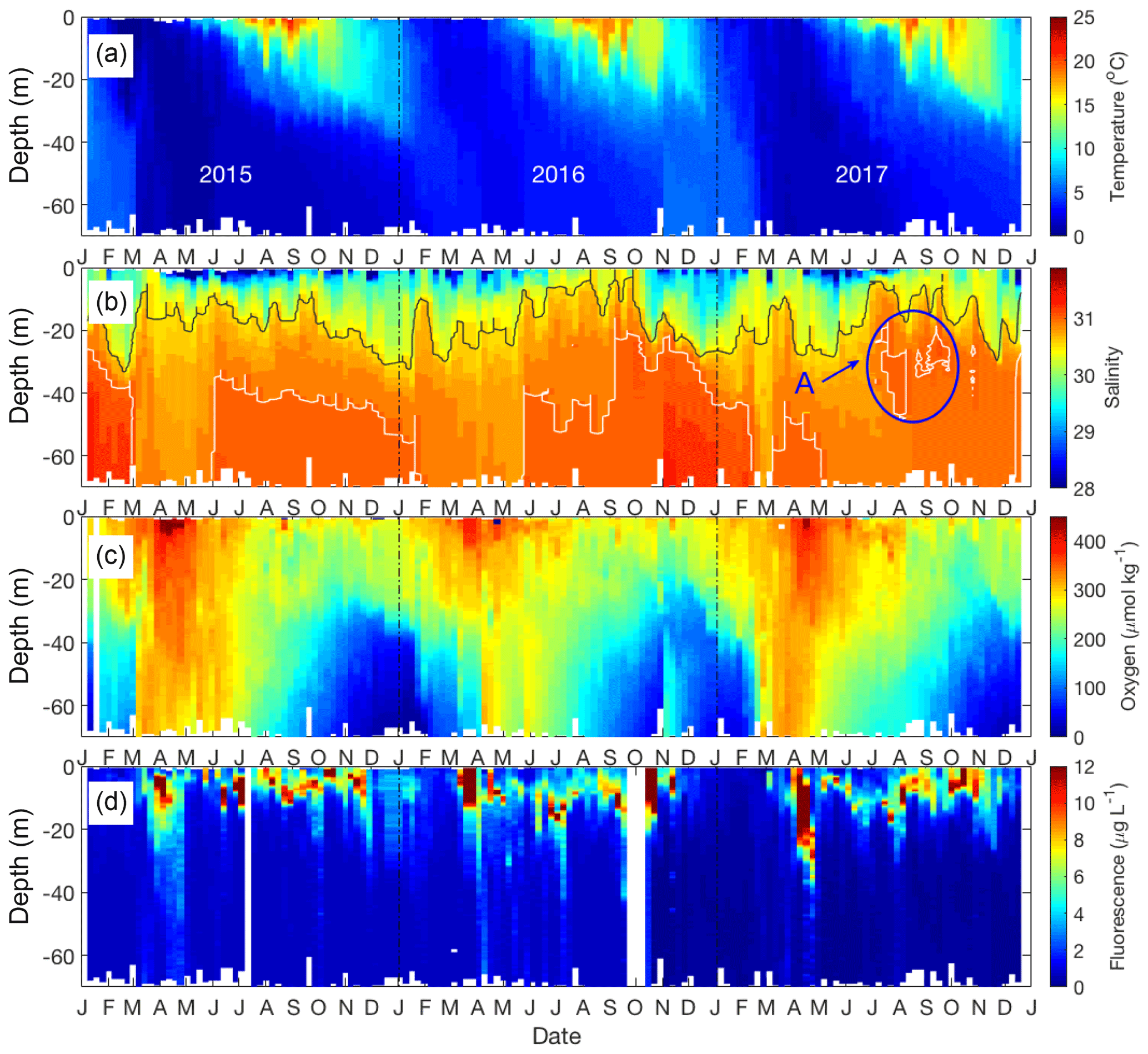
Figure 2Seasonal patterns of environmental and biological variables in Bedford Basin from January 2015 to December 2017. (a) Temperature; (b) salinity (grey contour line for S=30.5; white contour line for S=31). The blue circle (a) highlights a mid-depth intrusion. (c) Dissolved oxygen; (d) chlorophyll fluorescence.
3.1 Environmental variables from the Bedford Basin
The vertical profiles of temperature, salinity, dissolved oxygen and fluorescence (Fig. 2) are well mixed from top to bottom in late winter (February–March) as a result of wind mixing and convection (Li, 2001). Temperature is marked by strong seasonality to depths of <30 m. Near-surface temperatures start to rise above winter values of 4 ∘C, and stratified conditions develop around early April with temperatures reaching ca. 21 ∘C by the end of August (Fig. 2a).
Salinity ranges from 23 to 31 through the entire water column, with the lowest salinities occurring very close to the surface (Figs. 2b and 3c). The near-surface stratification varied both seasonally and between years, primarily in association with variability of precipitation and the discharge of the Sackville River (source: Environment and Climate Change Canada; http://climate.weather.gc.ca/climate_data/daily_data_e.html?StationID=50620&timeframe=2&StartYear=1840&EndYear=2018&Day=31&Year=2017&Month=10#, last access: November 2018). For example, the salinity difference between 1 and 5 m was >1 during much of the summer of 2015 (June to September) and summer 2017 (June to August). In summer 2016, however, the salinity at 1 m was close to that at 5 m (difference < 0.3) (Fig. 3c). Occasional intrusions of more dense water from the Scotian Shelf results in increased salinity, especially of bottom waters. The intrusions are irregular and tend to occur a few times per year, for instance in May 2016, at which time the salinity of bottom water increased from 30.8 to 31.0 (Fig. 2b), and in early July 2017 when the salinity of mid-depth water increased from 30.5 to 31 (see marked circle in Fig. 2b).
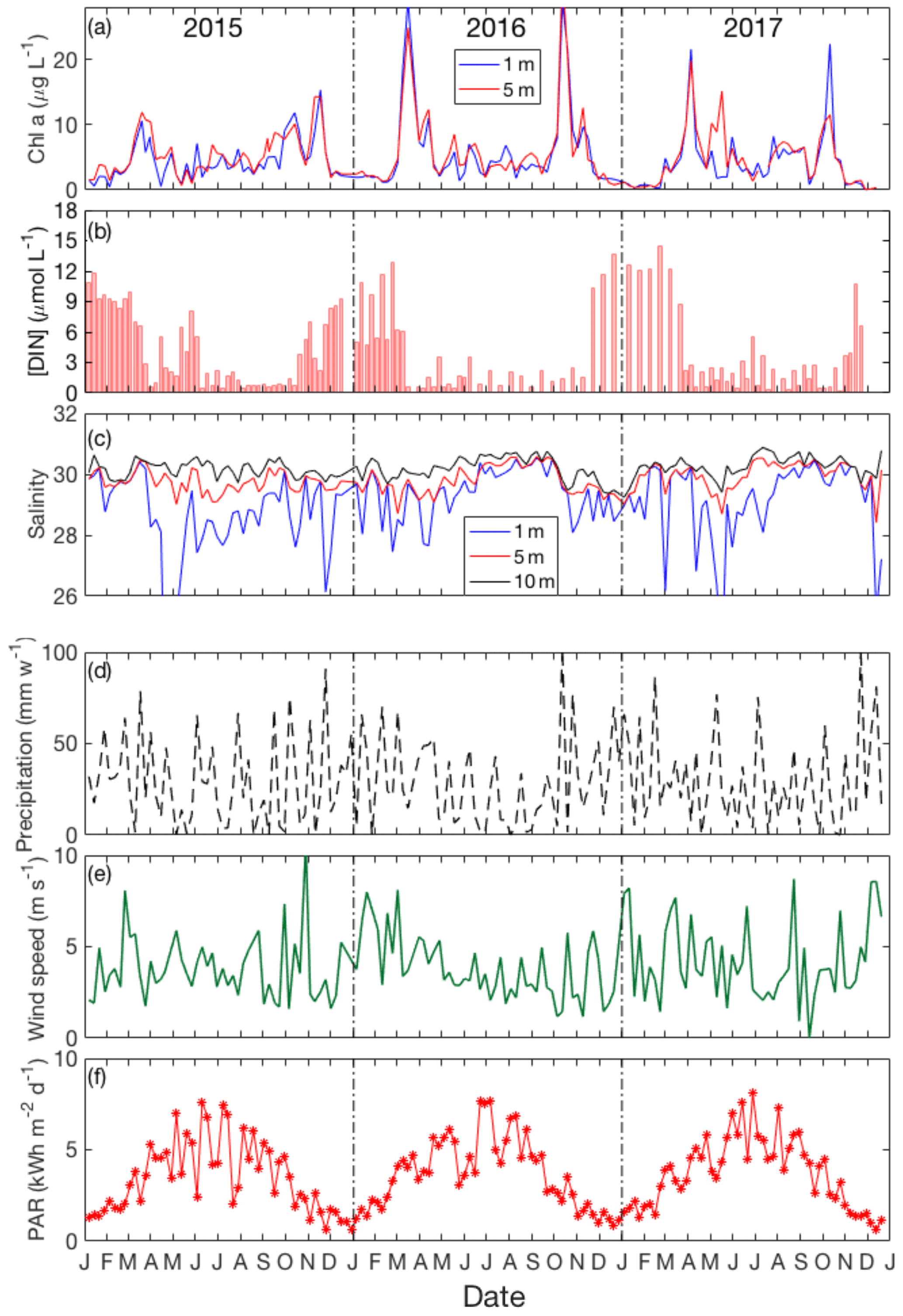
Figure 3Seasonal variation of (a) chlorophyll a: 1 m (blue line) and 5 m (red line); (b) DIN; (c) salinity in the surface layer: 1 m (blue line), 5 m (red line) and 10 m (black line); (d) weekly precipitation and (e) daily averaged wind speed (shown here only for sampling days); (f) weekly average PAR from January 2015 to December 2017.
The dissolved oxygen time series (Fig. 2c) shows the effect of temperature-dependent solubility variations in surface waters as well as intrusions and late winter vertical mixing in deeper water. In surface water the highest O2 concentrations occurred between March and April every year in association with the lowest seawater temperature. The vertical gradient of O2 concentration was generally smallest towards the end of April as a result of vertical mixing. Subsurface O2 concentrations (>30 m) generally decreased in summer due to respiration, with occasional interruptions of this O2 decline (e.g., November 2016) as a consequence of shelf-water intrusions, which brought sudden increases in O2 levels.
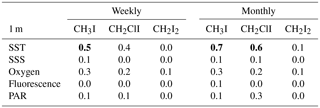
Table 1R2 value (Pearson's correlation coefficients, p<0.05) for the individual iodocarbon data based on both weekly data and monthly average. The significant correlations are highlighted with bold font.
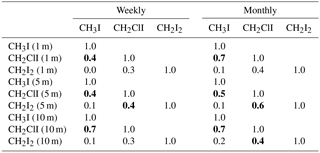
Figure 3 depicts time–depth plots of the variation of chlorophyll a, total dissolved inorganic nitrogen (DIN = [] + [] + []), salinity, precipitation, wind speed and solar irradiance in Bedford Basin over the period of the VOI sampling. The seasonal variations of chlorophyll a concentration in surface water (Fig. 3a) show that two blooms (spring and autumn) occur in surface water. For example, in 2016, chlorophyll a increased rapidly from March to April (from 5 to 26 µg L−1) and from September to October (from 10 to 28 µg L−1). The vertical variation of chlorophyll a (as determined from fluorescence measured on the CTD; see Fig. 2d) reached 12 µg L−1 during the bloom period. Subsurface (20–40 m) fluorescence-derived chlorophyll a dropped down to 4 µg L−1. In the near-bottom water chlorophyll a ranged between 0 and 2 µg L−1 during the whole year and varied only slightly.
The seasonal variation of dissolved inorganic nitrogen (DIN) in surface water is plotted in Fig. 3b. In winter, when chlorophyll a levels are very low due to light limitation, DIN concentrations reach ca. 12 µmol L−1 but are drawn down to low levels (<1 µmol L−1) after the spring bloom. Summertime chlorophyll a levels are moderate but variable (ca. 3 to 10 µg L−1), likely reflecting continuing nutrient input (e.g., from runoff and/or sewage treatment plants). The average precipitation in Bedford Basin was 27.6 mm week−1 in summer 2015 and 16 mm week−1 in summer 2016 (Fig. 3d). Typically, the strongest irradiance (data were downloaded from the CERES FLASHFLUX system: https://ceres-tool.larc.nasa.gov/ord-tool/jsp/FLASH_TISASelection.jsp, last access: November 2018) occurs in June and July (see Fig. 3f), and the highest water temperatures are observed in August.
3.2 Variations of iodocarbon concentrations in Bedford Basin
Iodocarbon concentrations in surface water (1, 5 and 10 m) showed strong seasonality, with the lowest concentrations from December through May (1.2 pmol L−1 for CH3I; 1.3 pmol L−1 for CH2ClI; 0.3 pmol L−1 for CH2I2). Concentrations start to increase in late May–June, reaching levels as high as 45 pmol L−1 for CH3I, 160 pmol L−1 for CH2ClI and ca. 80 pmol L−1 for CH2I2 (with a single peak of 500.5 pmol L−1; Fig. 4). Near-surface summertime concentrations of all three compounds were characterized by a broad seasonal peak 6–7 months in duration (or shorter for CH2I2), on top of which were superimposed three to four peaks of ca. 1 month duration. The number, amplitude and timing of these peaks varied amongst the three compounds with CH3I notably showing only one large peak in 2016 and four during the other 2 years of the time series (Fig. 4a).
Concentrations at 60 m were almost always lower and much less variable, ranging over the year from 1 to 9 pmol L−1 for CH3I (except fall–winter 2015–2016; see below), 1 to 6 pmol L−1 for CH2ClI and 0.4 to 18 pmol L−1 for CH2I2 (Fig. 4d). Hence, the bottom water (60 m) concentrations of CH2I2 and CH2ClI were always much lower than in near-surface waters throughout the summers. The surface to deep concentration difference was smallest for CH3I and showed interannual variability. Notably, bottom water concentrations reached 26 pmol L−1 and were even higher than in contemporary surface waters from September 2015 to March 2016 (Fig. 4d). Missing from the bottom water time series were the ca. 1 month duration variations seen in summertime surface water.
Interrelations between the iodocarbons in surface seawater were examined with linear regression of both weekly and monthly averaged concentrations. The resulting correlations are shown in Table 1. Using weekly data, significant correlations (i.e., p<0.05) were found between [CH3I] and [CH2ClI] at 1, 5 and 10 m depths with the strongest correlation (R=0.7) at 10 m. The only other significant correlation was between CH2I2 and CH2ClI at 5 m. Use of monthly averaged values gave stronger correlations. Once again, the significant correlations were between CH3I and CH2ClI (at 1, 5 and 10 m of depth) as well as between CH2ClI and CH2I2 at 5 and 10 m of depth. Table 2 also presents the correlations of iodocarbon concentrations with potentially related variables (discussed in Sect. 4.3).
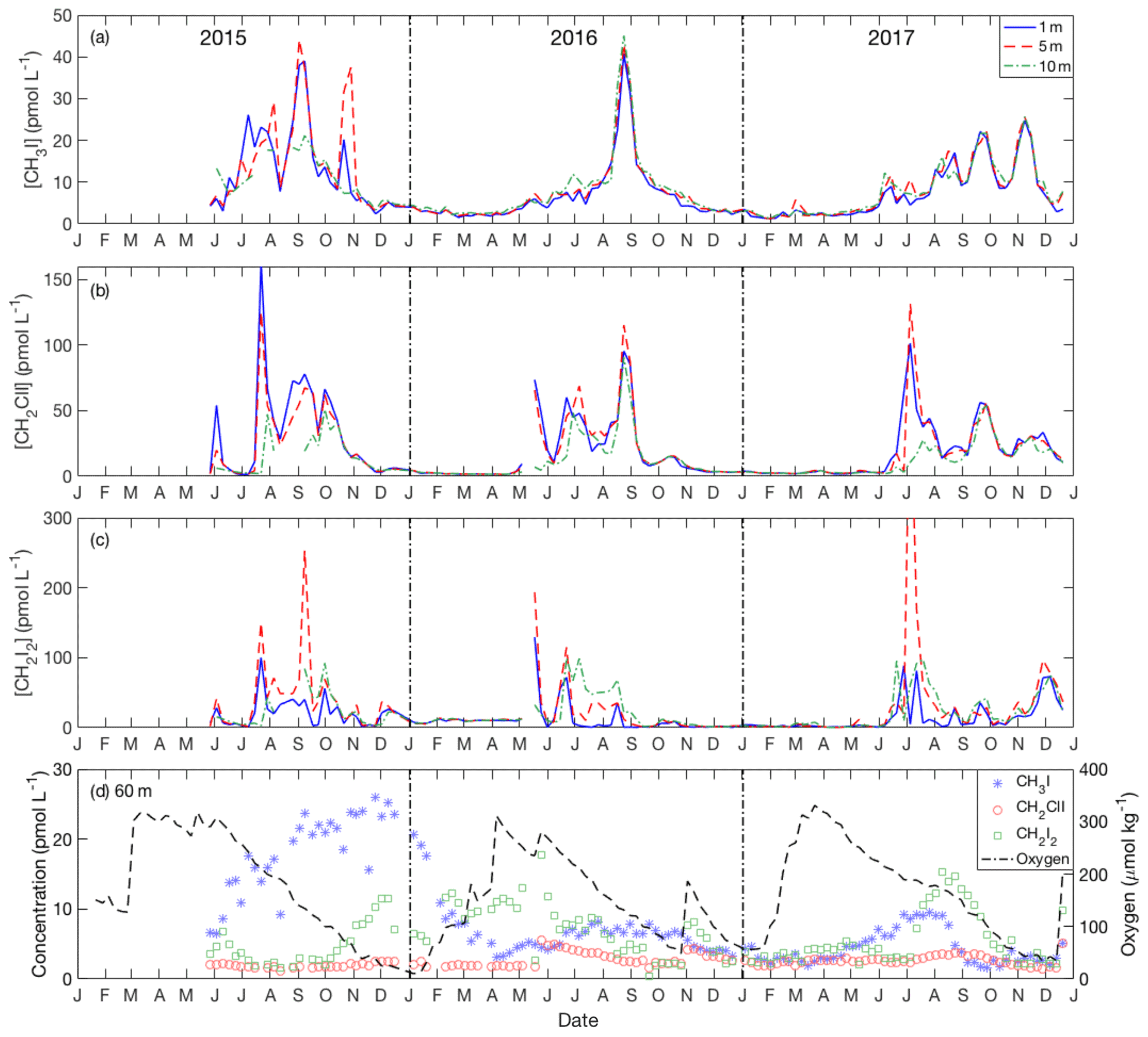
Figure 4Seasonal variation of iodocarbons in the Bedford Basin at 1 m (blue line), 5 m (red line) and 10 m (green line) from May 2015 to December 2017: (a) CH3I, (b) CH2ClI and (c) CH2I2. (d) Time series from near-bottom water (60 m) of iodocarbons and dissolved oxygen.
Table 2R2 value (Pearson's correlation coefficients, p<0.05) for iodocarbons and potentially relevant parameters based on both weekly data and monthly average. The significant correlations are highlighted with bold font.
Generally, the concentration of CH2I2 was higher than that of CH2ClI. The average ratio of CH2I2 ∕ CH2ClI within the top 10 m of the water column over the summer months was 1.4. However, this ratio was significantly lower at 1 m of depth (average of 0.6) and increased with depth (1.5 at 5 m and 2.2 at 10 m, reaching values as high as 2.7 at 60 m).
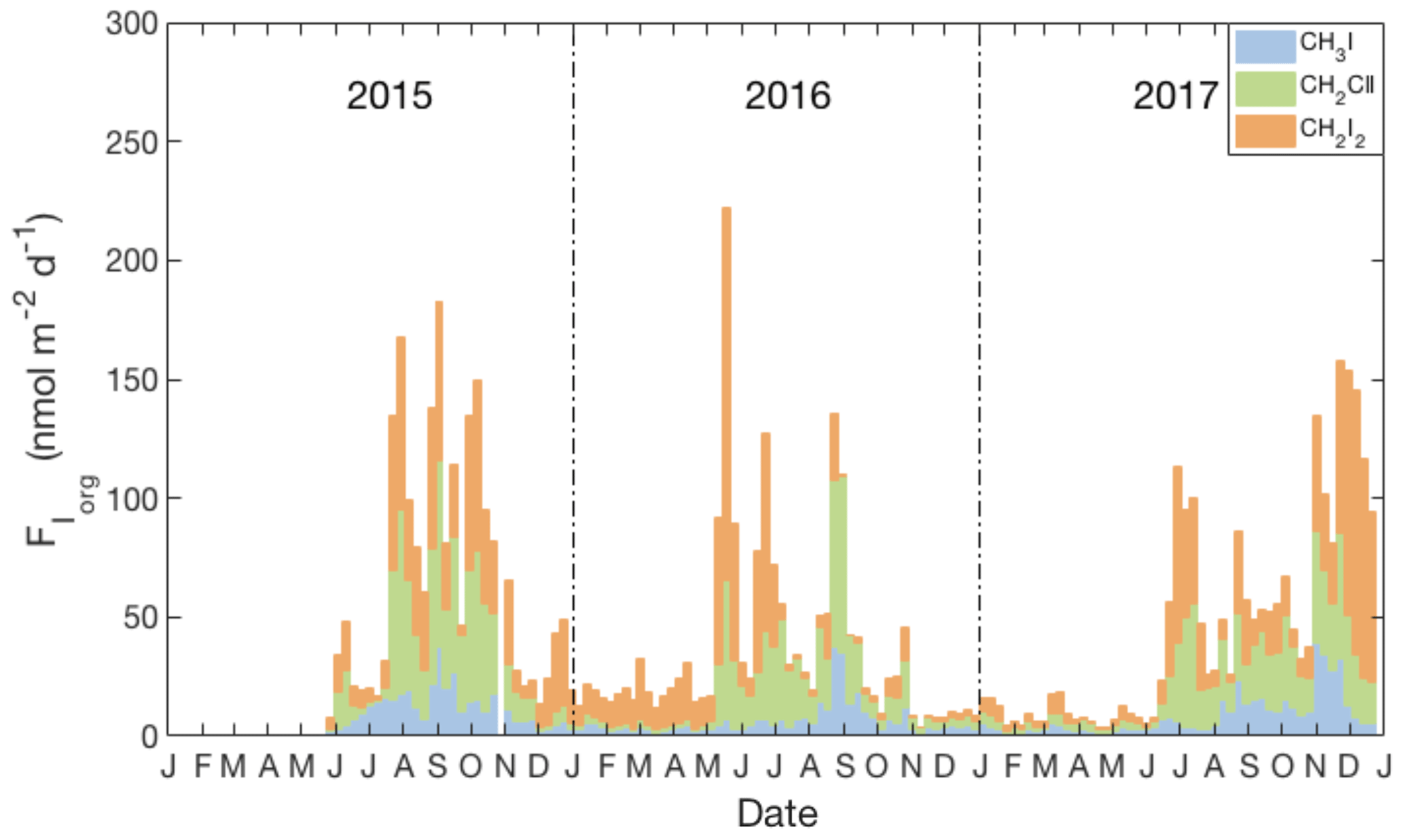
Figure 5Weekly averages of daily sea-to-air flux estimates of Iorg (see Sect. 3.3), including relative contributions of individual compounds (blue: CH3I, green: CH2ClI, orange: CH2I2) and using the parameterization for transfer velocity (kw) of Nightingale et al. (2000) and the air-side transfer velocity (ka) of Duce et al. (1991).
3.3 Sea-to-air flux
Using the concentrations of CH3I, CH2ClI and CH2I2 at 1 m of depth (Fig. 4) we estimated the sea-to-air flux of VOIs (F) using the following equation and the parameterization of Nightingale et al. (2000) for the waterside transfer velocity:
where u10 is wind speed at 10 m of height, Sc is the temperature-dependent Schmidt number, as estimated by Groszko (1999), and Henry's law constants (H) were from Moore et al. (1995). The air-side resistance has been shown by Archer et al. (2007) to be significant for soluble gases such as CH2ClI and CH2I2. Hence, K in Eq. (1) is calculated as follows (Liss and Slater, 1974):
where the air-side transfer velocity was calculated according to Duce et al. (1991).
with MW being the molecular weight of the gas of interest.
Daily averaged wind speed was measured at the nearby Halifax Dockyard (Fig. 1) (source: Environment and Climate Change Canada; http://climate.weather.gc.ca/index_e.html, last access: November 2018). Seawater temperature and surface iodocarbon concentrations were interpolated linearly between the weekly measurements in order to coincide with the wind speed data and generate daily flux estimates.
Following Archer et al. (2007) and Shimizu et al. (2017), we applied an atmospheric concentration of zero for calculating the flux of all three compounds. Rasmussen et al. (1982) reported an average atmospheric mixing ratio of CH3I of ca. 1 pptv for Cape Meares (45∘ N) and Yokouchi et al. (2008) presented a mean concentration of 0.98 pptv for Cape Ochiishi (43.2∘ N), with both sites sharing a similar latitude to our sampling location (44.69∘ N). If a mixing ratio of 1 pptv had been used for our calculations, the total annual flux of CH3I would have been reduced by only 5 %. Atmospheric mixing ratios of CH2ClI and CH2I2 are generally lower (reviewed by Carpenter, 2003) so that any overestimation of the fluxes of these compounds due to this assumption will certainly be negligible (Archer et al., 2007).
Table 3Seasonal variation of total sea-to-air fluxes of iodocarbons (nmol m−2 day−1). The highest flux values of iodocarbons in each year are marked in bold.
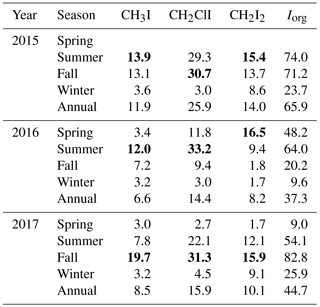
Figure 5 presents the weekly averaged combined flux of organically bound iodine, FIorg, as stacked bar charts, where . The calculated emissions of individual compounds ranged from 0.9 to 39.2 nmol m−2 day−1 for CH3I (annual average of 8.4 nmol m−2 day−1), 0.9 to 78.0 nmol m−2 day−1 for CH2ClI (annual average of 17.4 nmol m−2 day−1) and 0.3 to 78.0 nmol m−2 day−1 for CH2I2 (annual average of 10.3 nmol m−2 day−1). Seasonal and annual average fluxes of the individual compounds and of Iorg are presented in Table 3. Clearly, the sea-to-air flux is highest in summer and fall and is dominated by the flux of the dihalomethanes rather than CH3I.
3.4 Net production of CH3I
We used a mass balance approach to estimate the production rate of CH3I within the uppermost 10 m of the water column based on the concentration time series (see also Shi et al., 2014b). Using the annual cycle of average near-surface CH3I concentrations (Fig. 4a), we examined the mass balance of CH3I for this fixed depth interval according to
where ΔC is the daily change in the average CH3I concentration in the near-surface seawater (0–10 m); Pnet represents the net gross production minus any additional uncharacterized losses such as microbial degradation; Lsea-to-air is the sea-to-air flux (Sect. 3.3) and is the “chemical” loss due to nucleophilic substitution of Cl− for I−, which was calculated based on reaction kinetics (Elliott and Rowland, 1993; Jones and Carpenter, 2007) using the corresponding temperature, salinity and mean concentration of CH3I. averaged 0.1 pmol L−1 day−1. Lmix is the loss due to downward mixing and has been shown in several studies to be negligible compared with other loss terms (e.g., Richter and Wallace, 2004). The latter assumption will not always be valid in winter, especially when mixed layers deepen to >10 m. However, most production of iodocarbons occurs during summer and fall when there is strong stratification within the upper 10 m (see Fig. 3c), and we excluded from our mass balance calculations the limited periods of time when density was uniform in this depth interval.
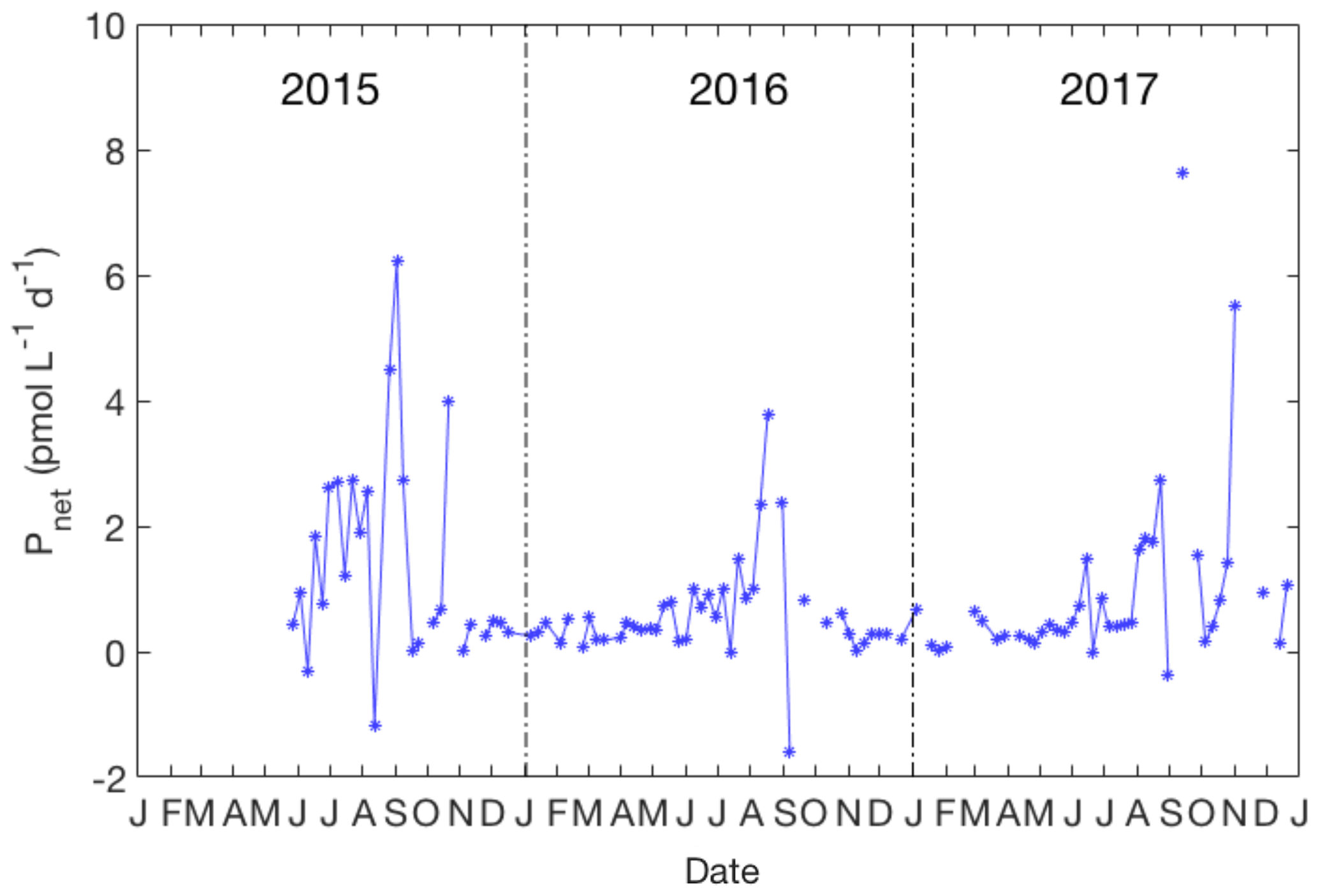
Figure 6Variation of the net production rate of CH3I in the upper 10 m from 2015 to 2017. No data are plotted when the layer of uniform density extended below 10 m.
The net production rate of CH3I over the 3-year time series is shown in Fig. 6. The annual average production rate of CH3I was 1.0 pmol L−1 day−1 (ranging from −1.6 to 8.5 pmol L−1 day−1). A significant peak of Pnet occurred from August to September in every year. The net production rate of CH3I in summer and fall averaged 1.6 pmol L−1 day−1 and was 5 times larger than wintertime rates (ca. 0.3 pmol L−1 day−1). Net production rates for CH2ICl and CH2I2 were also calculated (results not shown), with typical summertime values being 3.2 and 1.3 pmol L−1 day−1, respectively. Photolytic loss can be very significant for these compounds and has not been estimated, so these Pnet values represent the net gross production minus uncharacterized losses, including photolysis and microbial degradation.
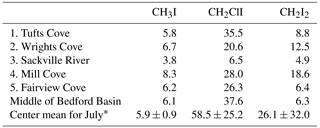
Table 4Concentration (pmol L−1) of iodocarbons measured at nearshore locations around Bedford Basin and at the regular sampling location on 19 July 2017 (1: Tufts Cove, 2: Wrights Cove, 3: Sackville River, 4: Mill Cove, 5: Fairview Cove) (see Fig. 1a). “Center” refers to the regular sampling site for the weekly time series. * Center mean for July: the average (and SD deviation) of measurements at the regular weekly sampling location during the month of July 2017 (n=4).
In the following we discuss the Bedford Basin data in comparison with other studies that have reported concentrations of multiple iodocarbons, especially those that have reported time series covering an annual cycle (see citations in the Introduction). All of these time series are from midlatitude (40–60∘ N) nearshore or continental shelf environments subject to strong seasonal variations of light, temperature and biological productivity. There are no reported time series of seawater concentrations from low latitudes.
4.1 Potential influence of nearshore and/or macroalgal sources
The potential of nearshore macroalgae to cause elevated coastal iodocarbon concentrations has been mentioned in a number of studies (Giese et al., 1999; Manley and de la Cuesta, 1997; Schall et al., 1994). We investigated this in July 2017 by sampling at five nearshore sites around Bedford Basin (Fig. 1) and comparing nearshore concentrations with values measured at the regular sampling site in the center of the basin (Table 4). The nearshore results were consistently within 1 standard deviation of mean concentrations of VOIs measured at the center of Bedford Basin during July, indicating no significant difference. Klick (1992) also compared measurements on samples collected directly over a rich bed of macroalgae with samples collected further away from direct contact with macroalgae: whereas they observed significantly higher concentrations of bromocarbons in proximity to the macroalgae, there was no difference observed for CH2I2 and CH2ClI. Shimizu et al. (2017) sampled a number of nearshore regions around Funka Bay, including rocky shores with extensive macroalgae, and also found concentrations to be similar at both nearshore and central bay locations. We therefore conclude that any direct impact of macroalgae on measured organoiodine levels is small, even in coastal regions, which lends strong support to the conclusion by Saiz-Lopez and Von Glasow (2012) that macroalgae are only a minor global source of these compounds to the atmosphere.

Figure 7Contribution of iodocarbons to total organic iodine (Iorg) in surface seawater from different regions and studies. 1: Klick (1992); 2: Orlikowska et al. (2015); 3: Jones et al. (2010); 4: Jones et al. (2009); 5: Carpenter et al. (2000); 6: Orlikowska and Schulz-Bull (2009); 7: Shimizu et al. (2017); 8: this study; 9: Archer et al. (2007); 10: Hepach et al. (2016); 11: Kurihara et al. (2010).
4.2 Concentrations and relative abundance of iodocarbon compounds
The average concentration of total volatile organic iodine Iorg (where Iorg = [CH3I] + [CH2ClI] + 2[CH2I2]) and the relative contributions of the different compounds to Iorg from this and other studies are shown in Fig. 7. The combined concentrations of the three iodocarbons are highest but also show the highest variability ([Iorg] = 25 to 281 pmol L−1) in summertime coastal waters (loosely defined here as within a few kilometers of land). Continental shelf waters have lower concentrations of Iorg averaging 32 pmol L−1, with open ocean waters having comparable or lower concentrations (average Iorg=17 pmol L−1). Despite the differences in Iorg concentration ranges evident for different regions in Fig. 7, a one-way ANOVA showed no significant differences between the means for the three regions. However, after pooling shelf and open ocean results to make only two populations, a t test did reveal a significant difference with coastal waters at the 95 % confidence level.
The distribution of Iorg is contrary to the global distribution of CH3I reported by Ziska et al. (2013), who noted a tendency for the open ocean to have higher concentrations than coastal waters (their definition of “coastal” was within 1∘ latitude or longitude of land and therefore much broader than ours). As noted by Ziska et al. (2013), this may reflect higher CH3I concentrations in tropical and subtropical open ocean waters, as their general pattern was reversed in the Northern Hemisphere. The coastal waters depicted in Fig. 7 are largely from midlatitudes in the Northern Hemisphere.
The relative contribution of the dihalomethanes to Iorg also appears to vary between regions, with the ratio of dihalomethane I to Iorg, ([CH2ClI] + 2 ⋅ [CH2I2]) ∕ [Iorg], averaging 0.71, 0.69 and 0.55 in coastal, shelf and open ocean waters, respectively. While an elevated contribution of dihalomethanes in coastal waters is consistent with the report by Jones et al. (2010), regional differences apparent in Fig. 7 are not significant at the 95 % confidence level when tested with a one-way ANOVA.
Klick (1992), Jones et al. (2010) and Shimizu et al. (2017) reported concentrations of volatile organic iodine in summertime coastal waters that are comparable to or higher than those observed in Bedford Basin (i.e., average Iorg concentrations > 100 pmol L−1). Our results from Bedford Basin correspond closely with concentrations and relative contributions reported by Shimizu et al. (2017) for coastal waters in Funka Bay, Japan (Fig. 7). In these coastal surface waters, the CH2I2 concentration and contribution was highest on average, followed by CH2ClI, and the lowest was CH3I. In open ocean waters, the relative contribution of [CH3I] to Iorg appears higher, reaching over 50 % in some cases (see Fig. 7), with the contribution of CH2I2 generally being lower in the open ocean than in coastal waters. However, once again, these apparent regional differences are not significant at the 95 % confidence level.
In laboratory studies, Fuse et al. (2003) demonstrated that relatively large amounts of CH2I2 and I2 together with smaller but still significant amounts of CH2ClI and CHI3 can be produced, presumably abiotically, in dark incubations of (filtered) spent culture media with suspended bacterial cells and added [I−]. The CH2I2 ∕ CH2ClI production ratio was ∼35 and no mono-iodinated CH3I was produced in these experiments. The implication was that dissolved organic compounds within spent media were key to the production of polyiodinated compounds. In the absence of spent culture media, additions of oxaloacetic acid also resulted in the formation of CH2I2 and CH2ClI (with a lower ratio of CH2I2 ∕ CH2ClI of ∼10), suggesting that organic acids may be a substrate for their formation. The mechanistic role of the suspended bacterial cells was not clear; however, they may have supplied the haloperoxidases required for oxidation of I− (see also Hill and Manley, 2009). Martino et al. (2009) alternatively demonstrated that oxidation of dissolved iodide to I2 and HOI by reaction with ozone (e.g., Garland et al., 1980) in filtered (0.2 µm) seawater containing natural levels of dissolved organic matter also resulted in the formation of polyiodinated compounds (CH2I2, CH2ClI and CHI3) with CH2I2 ∕ CH2ClI production ratios ranging from 2 to 4. They suggested that the yield of various iodocarbons depends on “the abundance and perhaps on the nature of the organic substrate”, which “can vary widely both temporally and spatially”. We could not, however, find any obvious relationship of near-surface iodocarbon concentrations with local measurements of atmospheric ozone near Bedford Basin (results not shown).
We therefore suggest that the higher levels of CH2I2 observed in coastal waters, including Bedford Basin, reflect a higher supply rate of HOI, I2 and/or the organic precursors suitable for the formation of polyiodinated compounds. Ultimately, the reduction of seawater iodate to reduced forms, such as iodide, likely supports the potential for organoiodine formation. Addition of iodide has been identified in short-term experiments as a source of reactive iodine (iodine atoms) that can stimulate the photochemical formation of CH3I (Moore and Zafiriou, 1994). However, Shi et al. (2014b) found no positive correlation of [I−] with seasonal CH3I production in a field study in Kiel Fjord, noting that background iodide levels may have always been sufficient to support the production of pM levels of CH3I. Indeed, there is no a priori reason to expect a positive correlation of [I−] with iodocarbon production if the supply of more reactive iodine species (iodine atoms, HOI and/or I2) is the key proximate control. The short-term formation rate of reactive species from a background reservoir of I−, and hence the production of iodocarbons, may depend more on the availability of haloperoxidases, various oxidants and/or temperature-dependent kinetics. The conversion of I− to these reactive species could even lead to inverse correlations between I− and iodocarbons.
Relatively small quantities of CH2ClI were produced in several of the experiments cited above, yet observations in Bedford Basin show average CH2I2 ∕ CH2ClI ratios of 1.4 in the top 10 m of the water column. Production ratios in these experiments vary, as noted above, but laboratory studies have also shown that photolysis of CH2I2 can be an important source of CH2ClI in surface waters with a yield of 25 % to 35 % (Jones and Carpenter, 2005; Martino et al., 2005). We observed significant correlation between [CH2I2] and [CH2ClI] at 5 and 10 m of depth (but not at 1 m) (Table 1), which is consistent with a fraction of the CH2I2 production being transformed photochemically (the lack of correlation at 1 m may be due to the very rapid photolysis). However, the correlation may also reflect the original production ratio of the individual compounds (which laboratory experiments suggest may be substrate dependent). In both cases, however, dissolved organic matter (DOM) quality and quantity (possibly associated with terrestrial supply) and/or an elevated supply of I− are likely to be underlying reasons for the high concentrations of dihalomethanes observed in Bedford Basin and other coastal waters.
4.3 Temporal variations of iodocarbons in near-surface water
The following discussion of temporal variability is separated into consideration of seasonal and interannual variations.
4.3.1 Seasonal variations
All of the reported iodocarbon time series showed strong seasonality, with minimum and sometimes undetectable concentrations in winter and higher concentrations in summer. Near-surface (0–10 m) concentrations of all three iodocarbons in Bedford Basin, including CH3I, remained low until mid-May to mid-June, with their subsequent increase coincident with initial warming of near-surface waters from wintertime minimum temperatures of ca. 1–2 ∘C (lag < 1 month; Fig. 8a). Hence, the initial appearance of all three iodocarbons occurred more than 3 months after the seasonal increase in solar radiation, ca. 1–2 months after the spring bloom (Fig. 8d), after near-surface nitrate had been drawn down to low levels (Fig. 8e) and almost coincident with the seasonal temperature increase (Fig. 8a).
In the western English Channel (Archer et al., 2007), a gradual increase in CH3I commenced in February, coincident with the seasonal increase in solar radiation. Summertime values remained high, with some higher-frequency variation, and then decreased in September–October. The increase in CH2ClI and CH2I2 started later, in April, more or less coincident with both the spring bloom and initiation of near-surface warming from a wintertime minimum temperature of ca. 8 ∘C. Summertime values of CH2ClI and CH2I2 showed periodic variations similar to those observed in Bedford Basin (Sect. 3.2).
The lower temporal resolution of the study in Funka Bay (Shimizu et al., 2017), with sampling only every 1 or 2 months, precluded a detailed examination of timing. A gradual increase in CH3I appeared to start in March, during or towards the end of the spring bloom when surface water temperatures were still close to their wintertime minimum of −1 to 2.5 ∘C. The seasonal increase in CH2I2 and CH2ClI occurred later (May–June) at a time of rising water temperatures and low nutrient levels with concentrations remaining elevated through the summer and decreasing to wintertime levels in October.
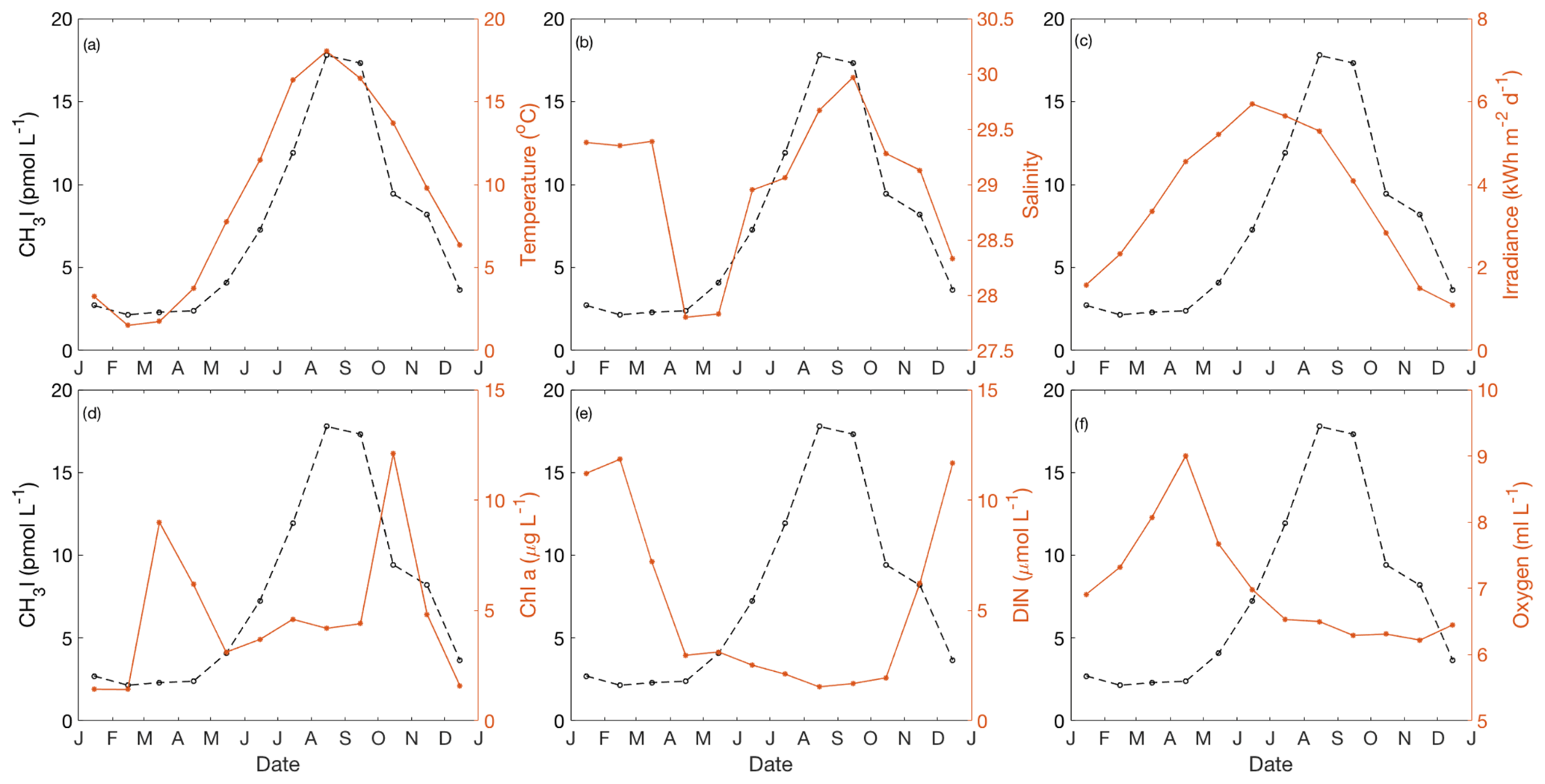
Figure 8Annual cycle of (a) temperature, (b) salinity, (c) irradiance, (d) Chl a, (e) dissolved inorganic nitrogen (DIN) and (f) dissolved oxygen for near-surface water (1–5 m) in Bedford Basin. The black dashed line depicts the annual cycle of CH3I. The figure presents the monthly mean values based on the data collected from May 2015 to December 2017.
The initial CH3I increase at a shallow station in the Kiel Fjord (Shi et al., 2014b) occurred in March and was closely linked in time to seasonal increases in solar radiation, temperature (winter minimum of 0 ∘C), Chl a and the springtime drawdown of nitrate. Lagged correlation analysis showed similarly strong correlations of CH3I with both temperature and solar radiation, with the annual cycle of CH3I lagging temperature by ca. 1 month; however, the very close correspondence of multiple seasonal cycles led the authors to note that “the use of correlation analysis to infer causality has likely reached its limit in this analysis”.
The observation of a rapid increase in the production rate of I− within phytoplankton cultures (diatoms and prymnesiophytes) when they enter stationary and especially senescent phases (Bluhm et al., 2011) is potentially relevant to the observed seasonality of iodocarbon formation. The reduction of iodate to iodide was suggested to be due to the release of precursors, such as reduced sulfur species, to a surrounding culture medium in association with a loss of membrane integrity by stressed cells or as a result of viral lysis. Hughes et al. (2011) also reported studies with cultures of Prochlorococcus marinas in which accumulation of CH3I commenced when cultures became senescent. We note that significant iodocarbon accumulation in Bedford Basin was confined to summertime when DIN was depleted (see Fig. 3b) and when cells may have been stressed or subject to viral lysis, perhaps similar to later stages of batch culture experiments.
We therefore hypothesize that seasonal nitrate drawdown leads to an increased supply of iodide to surface waters, which can in turn lead to the increased formation of iodine atoms, HOI and I2 as precursors for iodocarbon formation by both photochemical and haloform reaction pathways (Martino et al., 2009; Moore and Zafiriou, 1994). Whereas the supply of iodide may be one key control, it is likely that variations in light intensity and water temperature also contribute to the overall seasonality of the production rate of CH3I (e.g., through temperature influence on reaction kinetics). For example, light can influence the formation of CH3I directly (e.g., Moore and Zafiriou, 1994; Richter and Wallace, 2004). Light can also influence iodocarbon production indirectly, for example by producing oxidants such as H2O2 to promote oxidation of iodide by haloperoxidases (Hill and Manley, 2009) or by altering the quality of dissolved organic matter. The time series of CH2I2 and CH2ClI from very shallow (<4 m) nearshore waters of the Kattegat, Sweden (Klick, 1992), and the Baltic Sea, Germany (Orlikowska and Schulz-Bull, 2009), showed peaks in April–May and again in September–October, with low concentrations throughout summer. This contrasts with the deeper water columns of Bedford Basin, Funka Bay and the English Channel where concentrations remain elevated throughout summer. This likely reflects the dominance of photolytic loss over production within very shallow water columns exposed to summertime light intensities and long periods of daylight. Subsurface production coupled with vertical mixing may explain the summertime persistence in deeper water columns.
In addition to the broad seasonal variation, a number of maxima with durations of ca. 1 month were observed and appear similar to short-period fluctuations observed in the English Channel time series (Archer et al., 2007). There does not appear to be any consistent pattern linking the maxima and minima of the three compounds in the two studies, so the underlying causes for these shorter-period variations are unclear.
4.3.2 Interannual variability
The Bedford Basin time series is unique in having high-temporal-resolution sampling (weekly) over three annual cycles, which allows interannual variability to be examined for the first time. The most obvious interannual difference was in the behavior of CH3I. In particular, 2016 was markedly different in that only a single peak was observed in late August, whereas the summers of 2015 and 2017 were marked by three to four quasi-periodic, multi-week maxima. As noted already, the English Channel time series of CH3I did not exhibit this behavior. The shallow-water time series of CH3I in the Kiel Fjord and coastal Baltic Sea (Orlikowska and Schulz-Bull, 2009; Shi et al., 2014b) also did not exhibit this type of variability. Because the cause of the periodicity itself is not understood or explained, discussion of reasons for its interannual variation must be highly speculative. One clear difference of 2016 relative to the other 2 years was the lower summertime precipitation and associated lack of near-surface salinity stratification. The temporal behavior of CH3I in 2016 might therefore be related to altered near-surface mixing dynamics within Bedford Basin or, alternatively, to decreased delivery of key precursors (e.g., DOM) from land via rivers and wastewater.
4.4 Vertical distributions and subsurface temporal variability
Figure 4 shows the near-surface concentration variations of the VOIs. For CH3I, concentrations were almost always uniform between 1, 5 and 10 m. For CH2ClI, the concentrations at 1 and 5 m were usually very similar (average difference −4.1 %; median −2.5 %); however, concentrations at 10 m of depth were noticeably lower for periods of time. For CH2I2, the highest concentrations were observed at a depth of either 5 or 10 m, with concentrations at 5 m occasionally peaking at very high levels (e.g., 250–350 pmol L−1) for short periods (less than 1 week). Concentrations at 1 m were almost always lower than at 5 m, with the percentage of reduction relative to 5 m averaging 52 % in summer. Concentrations at 10 m, on the other hand, were generally the same as or higher than those measured at 5 m (with the exception of the previously mentioned short-lived peaks).
These results are consistent with earlier studies of vertical profiles in the open ocean (e.g., Moore and Tokarczyk, 1993; Yamamoto et al., 2001) and with model predictions (Jones et al., 2010; Martino et al., 2006). In particular, our results are consistent with the quantitative predictions of a mixed-layer model (Jones et al., 2010) that CH2ClI would typically be nearly uniform within the upper 6 m of the water column, whereas photolytic decay could remove up to 100 % of the CH2I2 over that depth range, depending on time of day and conditions.
4.5 Temporal variability in near-bottom water (60 m)
The time series of VOIs in near-bottom waters (60 m) are presented in detail in Fig. 9a–c, with specific events labeled 1 through 9. Variability was generally of lower amplitude than in surface waters, except for CH3I during the winter of 2015–2016 (Fig. 4d). From June to December 2015, [CH3I] increased steadily (concentration change, ΔC=20 pmol L−1) (event 1 to 3; Fig. 9a), exceeding surface water concentrations from October 2015 until the end of March 2016. No comparable increase was observed during 2016 (Fig. 9b), and a smaller increase (ΔC=8 pmol L−1) was confined to the early summer of 2017 (Fig. 9c).
Concentrations of CH2ClI remained almost constant at <5 pmol L−1 throughout, with the notable exception of abrupt (<1 week) increases in May and November 2016 (events 4 and 5; Fig. 9b) and December 2017 (event 9; Fig. 9c). These increases (ΔC=2–5 pmol L−1) coincided with sudden increases in salinity (and O2) and reflect intrusion of saltier near-surface waters from offshore rather than local production. The subsequent concentration declines reflect loss due to mixing or, more likely, reaction and/or microbial degradation within the water column and sediments. The same three intrusions also drove abrupt increases in CH2I2 with amplitudes ca. 1.5–2 times higher than those for CH2ClI, consistent with near-surface concentration ratios (see Sect. 3.2). However, CH2I2 also showed higher-amplitude variations unrelated to the bottom water intrusions (see below).
The increase in CH3I from June through October 2015 (event 1 through 3; Fig. 9a) paralleled a steady decline in oxygen, suggesting that production was linked to the degradation of organic matter. The accumulation rate of ca. 0.06 pmol L−1 day−1 was 20 times smaller than typical Pnet for CH3I in surface waters (see Sect. 3.4). The increase appears consistent with results from short-term (3-day) incubation experiments with biogenic marine aggregates reported by Hughes et al. (2008) in which concentrations of mono-iodinated iodocarbons, including CH3I, increased but with no corresponding increase in dihalogenated compounds. Their results suggested the alkylation of inorganic iodine or the breakdown of higher-molecular-mass organohalogens as production pathways, and following Amachi et al. (2001), they suggested that microbial degradation increased the supply of precursors.
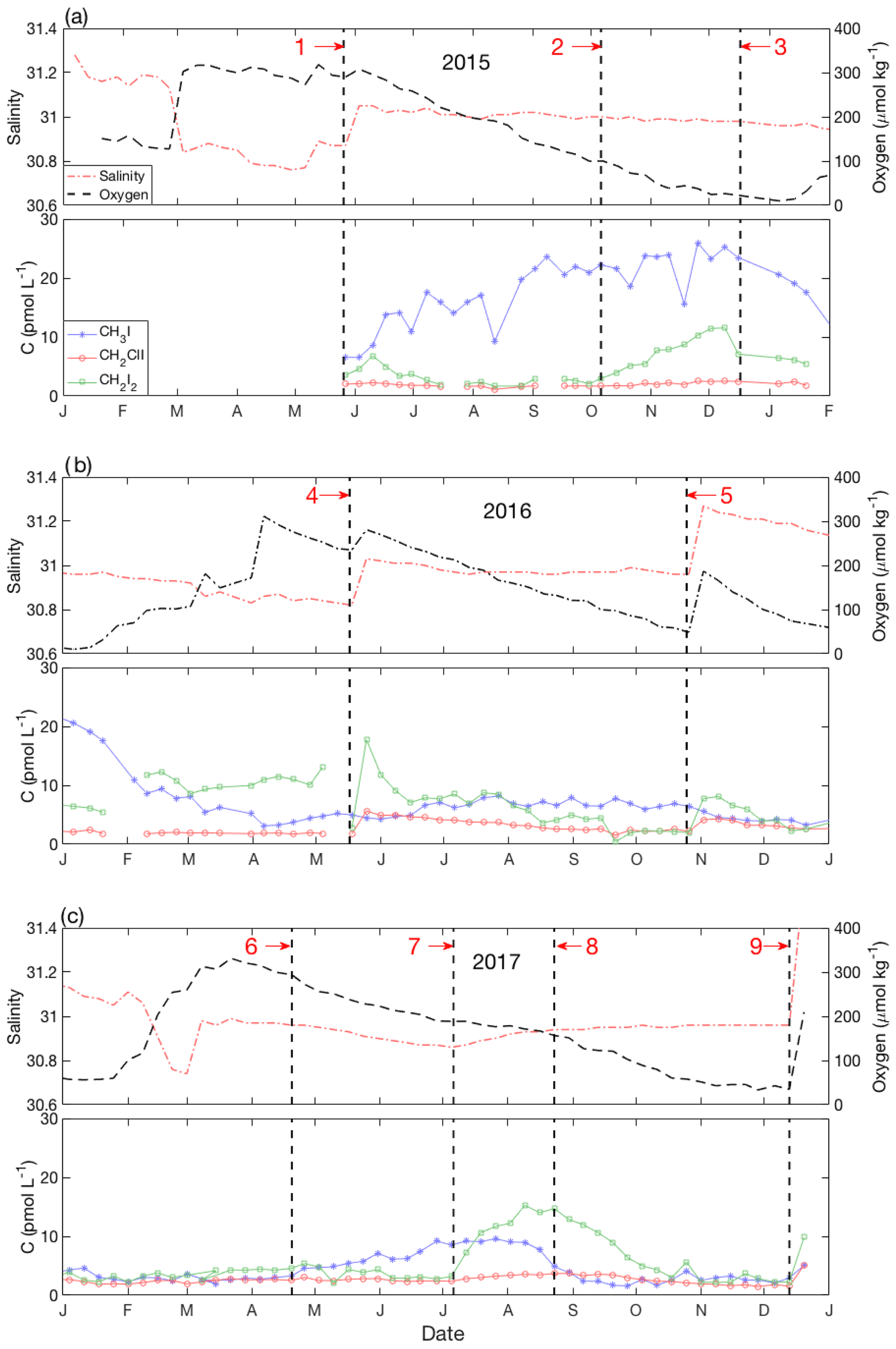
Figure 9Detailed time series from near-bottom waters in (a) 2015, (b) 2016 and (c) 2017. For each year, panel (a) shows the variability of salinity (red dash-dot line) and dissolved oxygen (black dashed line); panel (b) shows iodocarbons (CH3I: blue stars; CH2ClI: red open circles; CH2I2: green open squares). The vertical bold dashed lines (1 to 9) represent the special events discussed in Sect. 4.5.
However, as O2 concentrations declined further from October through late December (event 2 to 3; Fig. 9a), the concentration of CH3I stabilized and the CH2I2 concentration increased markedly from 2 to 12 pmol L−1. From January through April 2016, CH3I levels decreased (following event 3; Fig. 9a and b) in concert with increasing O2 concentrations and decreasing salinity, reflecting the effects of progressive vertical mixing with overlying waters that had lower CH3I concentrations. Over this same period, CH2I2 concentrations at 60 m remained almost constant (Fig. 9a and b) due to the smaller vertical concentration gradient until increasing due to an intrusion (event 4). This was followed by a gradual decrease over summer months in parallel with the seasonal decrease in O2 (Fig. 9b).
The data from 2015 and 2017 are suggestive of a “switch” of production mechanism from an alkylation pathway producing mono-iodinated compounds (CH3I) to a haloform-type reaction producing CH2I2. The switch took place in October 2015 (event 2; Fig. 9a) when oxygen concentrations dropped below 90 µmol kg−1, although whether the switch was related to redox conditions in the water column, sediments, speciation and availability of iodine, or availability of suitable organic precursors and/or enzymes cannot be determined.
There was almost no near-bottom iodocarbon production during 2016 (Fig. 9b), and therefore no switch, for reasons that are not clear given that O2 declined through summer and fall until interrupted by an intrusion in November (event 5; Fig. 9b). We speculate that the lack of production in near-bottom water might be linked to the relatively short period of CH3I production in near-surface waters during that year (Fig. 4a).
In 2017, there was moderate subsurface production of CH3I associated with O2 consumption (April through July; event 6 to 7) and, again, an apparent switch to CH2I2 production marked by plateauing of CH3I concentrations (event 7) at the same time as CH2I2 concentrations started to increase (Fig. 9c). This was followed by a subsequent decrease to background levels over a period of about a month. The apparent switch in production took place earlier in the year and at higher O2 concentrations (175 µmol kg−1) than in 2015.
However, close inspection of Fig. 2 shows that the plateauing of CH3I in near-bottom waters (event 7) also coincided with the appearance of a mid-depth intrusion of saltier water (as denoted by the 31 salinity contour; see blue circled area in Fig. 2). The same period (between events 7 and 8) was marked at 60 m by a change from declining to increasing salinity, reduction in the rate of oxygen concentration decline (Fig. 9c) and an increase in the rate of warming (not shown). The mid-depth salinity maximum in Bedford Basin must reflect the intrusion of saltier water from offshore. The subsequent trends in temperature, salinity and dissolved oxygen at 60 m between events 7 and 8 (Fig. 9c) are consistent with mixing of preexisting near-bottom water with this intrusion. It therefore cannot be ruled out that mixing with this intrusion contributed in some way to the near-bottom increase in CH2I2, plateauing of CH3I concentrations, the small but significant increase in CH2ClI (ΔC=1–2 pmol L−1) and hence to the apparent switch, which all occurred at the same time.
At the end of this period in mid-August (event 8), the rate of warming and salinity increase at 60 m decreased again and the rate of oxygen decline increased (Fig. 9c), suggesting that the intrusion's impact had lessened. At this time, CH2I2 and CH3I concentrations started to return to background levels with an estimated half-life, assuming first-order kinetics, of ca. 65 days (CH2I2) and 14 days (CH3I). A decrease in [CH2ClI] started about a month later with a first-order half-life of ca. 70 days. Similar rates of decline of [CH2ClI] and [CH2I2] were observed following sudden concentration increases associated with other intrusions, as discussed earlier.
This very detailed discussion of temporal variability emphasizes that a variety of physical and biogeochemical mechanisms can contribute to interannual, seasonal and short-term variability of the three iodocarbons. The high-amplitude variability observed in Bedford Basin could prove useful for the validation of models representing complex iodine cycling and physical mixing dynamics. However, separation of multiple potential contributing factors and processes underlying temporal variability may require a dataset with higher vertical resolution and, ideally, a seasonally resolved time series of experiments (compare Shi et al., 2014a).
4.6 Sea-to-air fluxes
The temporal variation of the sea-to-air flux of Iorg and the relative contribution from the three iodocarbons are shown in Fig. 5 and Table 3. Similar to the findings of Archer et al. (2007), air-side resistance leads to significant reductions in calculated annual average fluxes for CH2ClI and CH2I2 of 10 % and 24 %, respectively, relative to calculations when it is ignored. The study by Shimizu et al. (2017) did not consider air-side resistance, so Table 5 presents both their original reported fluxes and fluxes adjusted for its likely impact based on our study and that of Archer et al. (2007). The following discussion makes use of the adjusted fluxes.
Consistent with earlier time series (excluding those from very shallow waters; see Sect. 4.2), the sea-to-air flux of iodocarbons is generally highest in summer–fall. However, high wintertime fluxes are also possible, as seen in 2017 when there was a large efflux of CH2I2 (averaging 9.1 nmol m−2 day−1; Table 3) due to both strong winds and relatively high concentrations. The fluxes of CH3I and CH2ClI, on the other hand, were always higher in summer–fall (ca. 3–5 times and 10 times higher, respectively). Similar findings were presented by Shimizu et al. (2017) with the total iodine flux in Funka Bay in summer being >4 times that in winter.
Table 5Comparison of sea-to-air flux (nmol m−2 day−1) of total organic iodine from different studies. English Channel is an average for 1 year; Funka Bay value is averaged over 3 years; Kiel Fjord is averaged over 2 years but for CH3I only; Bedford Basin (this study) is an average over 3 years. Seasons are as defined in this study (see Sect. 2). For Funka Bay, values in parentheses represent fluxes that have been adjusted from the original reported values to take into account the effect of air-side resistance using correction factors of 12 % and 28 % for H2ClI and CH2I2, respectively (based on average effects reported in Archer et al. (2007) and this study; see Sect. 4.6).
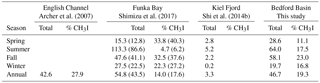
Our estimated emissions of CH3I (8.4 nmol m−2 day−1, Table 5) are in the range calculated previously for coastal and continental shelf water in similar latitudes (11.9 and 7.7 nmol m−2 day−1; Archer et al., 2007; Shimizu et al., 2017, respectively). The average flux of CH3I reported by Jones et al. (2010) from the west of Ireland was 4 times higher but based on a sampling period of only 1 month during summer. Sea-to-air fluxes of CH2ClI from Funka Bay and the English Channel were similar to our calculated fluxes from Bedford Basin. However, the highest variation is observed in the annual averaged flux of CH2I2, ranging from 3.5 nmol m−2 day−1 (the west English Channel) to 10.3 nmol m−2 day−1 (Bedford Basin) and 12.6 nmol m−2 day−1 (Funka Bay, Japan). The total annual Iorg sea-to-air flux from Bedford Basin averaged 46.7 nmol m−2 day−1, which was approximately 5 times larger than the flux due to CH3I alone. The total annual flux was similar between all three locations.
Figure 5 and Table 3 show that the total Iorg flux is subject to significant interannual variability, which could not be assessed by earlier studies. Notably, the Iorg flux in 2016 was ca. 2 times smaller than in 2015 and 2017. A comparison of wind speeds and concentrations showed that the influence of wind speed was dominant due to winds during summer–fall of 2016 being 1–2 m s−1 lower.
4.7 Production rate of CH3I
The annual mean production rate (Pnet) of CH3I in this study, estimated using Eq. (1), was 1.0 pmol L−1 day−1 (ranging from −1.6 to 8.5 pmol L−1 day−1; see Sect. 3.4 and Fig. 6). This is comparable with the global average production rate estimated by Stemmler et al. (2013) (1.64 pmol L−1 day−1), for which 70 % was produced via a photochemical mechanism. Based on data presented by Archer et al. (2007), the annual mean production rate of CH3I in the western English Channel was ca. 2 pmol L−1 day−1 (range: −0.2 to 6 pmol L−1 day−1). Here it should be noted that their “minimum gross production rate” is equivalent to Pnet in this study and in Shi et al. (2014a). In contrast, Shi et al. (2014b) estimated a considerably lower annual mean net production rate in the Kiel Fjord of ca. 0.1 pmol L−1 day−1 (maximum of 0.8 pmol L−1 day−1). The maximum production rates from the Kiel Fjord study were smaller as they were based on monthly average (and therefore “smoothed”) concentrations. However, Shi et al. (2014a) also conducted weekly incubation experiments, which gave in vitro values of Pnet that were closely comparable with the field-based estimates in Kiel Fjord.
The lower values of Pnet in the Kiel Fjord compared with both Bedford Basin and the English Channel must reflect either differences in gross production (e.g., due to differences in the supply of precursors and reactants such as iodide) or differences in other uncharacterized losses. Evidence of a poorly characterized loss process, possibly microbial degradation, was in fact observed in the Kiel Fjord incubation experiments (Shi et al., 2014a). On the other hand, incubation experiments conducted with additions of labeled methyl iodide (13CD3I) to Bedford Basin surface waters (data not shown) during the course of this study showed no such losses. We therefore hypothesize that the lower Pnet in Kiel Fjord is a result of higher microbial degradation of CH3I in that very shallow (<12 m) nearshore environment.
The 3-year time series of weekly iodocarbon concentrations from Bedford Basin shows overall seasonality similar to that observed in coastal time series from both the English Channel and Funka Bay, Japan. There was no midsummer minimum in the concentration of polyiodinated compounds as observed in some time series from very shallow water (<10 m), which likely reflects the dominance of photolytic decay in such shallow water columns. Interannual variability in near-surface water concentrations was particularly pronounced for CH3I, with only a single short-lived concentration maximum observed in 2016, possibly as a result of anomalously low rainfall and a consequently reduced supply of terrestrial organic matter during that summer.
Based on the time series and published lab studies, we hypothesize that seasonal near-surface production of iodocarbons is linked to accelerated reduction of iodate to iodide under post-bloom conditions following the disappearance of nutrients and possibly also influenced by water temperature. The observed vertical variation of CH2I2 and CH2ClI is consistent with more rapid photolysis of CH2I2.
The average annual sea-to-air flux of total volatile organic iodine (46.7 nmol m−2 day−1) is almost identical to that observed in Funka Bay, Japan, and the English Channel. The polyiodinated compounds contributed ca. 80 % of the total flux, which was similar to that in the other two time series and confirms that the sea-to-air flux of polyiodinated compounds dominates in coastal waters. The fluxes were variable on interannual timescales (factor of 2) as a result mainly of wind speed variability.
The near-bottom water (60 m) time series was impacted by episodic intrusions of water from offshore and showed evidence of CH3I production associated with the decay of organic matter, albeit with a production rate more than an order of magnitude lower than in surface waters. The time series showed evidence of a possible switch from CH3I production (e.g., by alkylation of organic matter) to the production of CH2I2 (e.g., by a haloform-type reaction) after periods of about 1 month.
The very high-amplitude concentration variations encountered in Bedford Basin, coupled with its relative accessibility for high-frequency sampling and constrained yet variable physical exchanges, make Bedford Basin a useful location to investigate iodine cycling. To date, the complexity of iodine biogeochemistry has hindered progress towards understanding the controls on spatial and temporal fluxes of iodine between the ocean and atmosphere. We suggest that progress can now be made through more comprehensive sampling (higher vertical resolution and inclusion of inorganic iodine speciation measurement) coupled with a biogeochemical model of Bedford Basin that includes iodine chemistry (e.g., Stemmler et al., 2013) and a time series of experimental studies conducted in the context of the time series (see Shi et al., 2014a). In other words, Bedford Basin may provide an ideal location and time series upon which to base a multi-investigator campaign to understand environmental controls on volatile iodine cycling and improve its representation in models.
The datasets generated during the current study are available in the Mendeley repository: https://data.mendeley.com/datasets/5y2t4mkjbv/1 (Shi and Wallace, 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/os-14-1385-2018-supplement.
QS and DW contributed to the design and implementation of the research, the analysis of the results, and writing the paper.
The authors declare that they have no conflict of interest.
This work was funded by the Canada Excellence Research Chair in Ocean
Science and Technology at Dalhousie University. Sampling of the Bedford
Basin time series was supported by MEOPAR Observation Core, Department of
Fisheries and Ocean, Canada (DFO), and the Bedford Basin Monitoring Program (BBMP)
(http://www.bio.gc.ca/science/monitoring-monitorage/bbmp-pobb/bbmp-pobb-en.php, last access: November 2018).
The authors thank Richard Davis, Anna Haverstock and the crew of the
Sigma T. Assistance and guidance in the laboratory from Claire Normandeau
and Liz Kerrigan are also acknowledged.
Edited by: Mario Hoppema
Reviewed by: two anonymous referees
Amachi, S., Kamagata, Y., Kanagawa, T., and Muramatsu, Y.: Bacteria mediate methylation of iodine in marine and terrestrial environments, Appl. Environ. Microbiol., 67, 2718–2722, 2001.
Archer, S. D., Goldson, L. E., Liddicoat, M. I., Cummings, D. G., and Nightingale, P. D.: Marked seasonality in the concentrations and sea-to-air flux of volatile iodocarbon compounds in the western English Channel, J. Geophys. Res.-Oceans, 112, C08009, https://doi.org/10.1029/2006JC003963, 2007.
Bluhm, K., Croot, P. L., Huhn, O., Rohardt, G., and Lochte, K.: Distribution of iodide and iodate in the Atlantic sector of the southern ocean during austral summer, Deep-Sea Res. Pt. II, 58, 2733–2748, 2011.
Brownell, D. K., Moore, R. M., and Cullen, J. J.: Production of methyl halides by Prochlorococcus and Synechococcus, Global Biogeochem. Cy., 24, GB2002, https://doi.org/10.1029/2009GB003671, 2010.
Buckley, D. E. and Winters, G. V: Geochemical characteristics of contaminated surficial sediments in Halifax Harbour: impact of waste discharge, Can. J. Earth Sci., 29, 2617–2639, 1992.
Burt, W. J., Thomas, H., Fennel, K., and Horne, E.: Sediment-water column fluxes of carbon, oxygen and nutrients in Bedford Basin, Nova Scotia, inferred from 224Ra measurements, Biogeosciences, 10, 53–66, https://doi.org/10.5194/bg-10-53-2013, 2013.
Carpenter, L. J.: Iodine in the marine boundary layer, Chem. Rev., 103, 4953–4962, 2003.
Carpenter, L. J., Malin, G., and Liss, P. S.: Novel biogenic iodine-containing trihalomethanes and other, Global Biogeochem. Cy., 14, 1191–1204, 2000.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, 2013.
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B. D., Hossaini, R., Laube, J. C., Yvon-Lewis, S. A., Engel, A., and Montzka, S. A.: Update on ozone-depleting substances (ODSs) and other gases of interest to the Montreal protocol, in Scientific assessment of ozone depletion: 2014, World Meteorological Organization, Geneva, 1.1–1.101, 2014.
Davis, D., Crawford, J., Liu, S., McKeen, S., Bandy, A., Thornton, D., Rowland, F., and Blake, D.: Potential impact of iodine on tropospheric levels of ozone and other critical oxidants, J. Geophys. Res.-Atmos., 101, 2135–2147, 1996.
Duce, R. A., Liss, P. S., Merrill, J. T., Atlas, E. L., Buat-Menard, P., Hicks, B. B., Miller, J. M., Prospero, J. M., Arimoto, R., Church, T. M., Ellis, W., Galloway, J. N., Hansen, L., Jickells, T. D., Knap, A. H., Reinhardt, K. H., Schneider, B., Soudine, A., Tokos, J. J., Tsunogai, S., Wollast, R., and Zhou, M.: The atmospheric input of trace species to the world ocean, Global Biogeochem. Cy., 5, 193–259, https://doi.org/10.1029/91GB01778, 1991.
Elliott, S. and Rowland, F. S.: Nucleophilic substitution rates and solubilities for methyl halides in seawater, Geophys. Res. Lett., 20, 1043–1046, 1993.
Fuse, H., Inoue, H., Murakami, K., Takimura, O., and Yamaoka, Y.: Production of free and organic iodine by Roseovarius spp., FEMS Microbiol. Lett., 229, 189–194, https://doi.org/10.1016/S0378-1097(03)00839-5, 2003.
Garland, J. A., Elzerman, A. W., and Penkett, S. A.: The mechanism for dry deposition of ozone to seawater surfaces, J. Geophys. Res.-Oceans, 85, 7488–7492, 1980.
Giese, B., Laturnus, F., Adams, F. C., and Wiencke, C.: Release of Volatile Iodinated C1–C4 Hydrocarbons by Marine Macroalgae from Various Climate Zones, Environ. Sci. Technol., 33, 2432–2439, 1999.
Groszko, W. M.: An estimate of the global air-sea flux of methyl chloride, methyl bromide, and methyl iodide, PhD Thesis, Dalhousie University, Dalhousie, 1999.
Hepach, H., Quack, B., Tegtmeier, S., Engel, A., Bracher, A., Fuhlbrügge, S., Galgani, L., Atlas, E. L., Lampel, J., Frieß, U., and Krüger, K.: Biogenic halocarbons from the Peruvian upwelling region as tropospheric halogen source, Atmos. Chem. Phys., 16, 12219–12237, https://doi.org/10.5194/acp-16-12219-2016, 2016.
Hill, V. L. and Manley, S. L.: Release of reactive bromine and iodine from diatoms and its possible role in halogen transfer in polar and tropical oceans, Limnol. Oceanogr., 54, 812–822, https://doi.org/10.4319/lo.2009.54.3.0812, 2009.
Hughes, C., Malin, G., Turley, C. M., Keely, B. J., and Nightingale, P. D.: The production of volatile iodocarbons by biogenic marine aggregates, Limnol. Oceanogr., 53, 867–872, 2008.
Hughes, C., Franklin, D. J., and Malin, G.: Iodomethane production by two important marine cyanobacteria: Prochlorococcus marinus (CCMP 2389) and Synechococcus sp. (CCMP 2370), Mar. Chem., 125, 19–25, 2011.
Jones, C. E. and Carpenter, L. J.: Solar photolysis of CH2I2, CH2ICl, and CH2IBr in water, saltwater, and seawater, Environ. Sci. Technol., 39, 6130–6137, 2005.
Jones, C. E. and Carpenter, L. J.: Chemical destruction of CH3I, C2H5I, 1-C3H7I, and 2-C3H7I in saltwater, Geophys. Res. Lett., 34, 1–6, https://doi.org/10.1029/2007GL029775, 2007.
Jones, C. E., Hornsby, K. E., Dunk, R. M., Leigh, R. J., and Carpenter, L. J.: Coastal measurements of short-lived reactive iodocarbons and bromocarbons at Roscoff, Brittany during the RHaMBLe campaign, Atmos. Chem. Phys., 9, 8757–8769, https://doi.org/10.5194/acp-9-8757-2009, 2009.
Jones, C. E., Hornsby, K. E., Sommariva, R., Dunk, R. M., Von Glasow, R., McFiggans, G., and Carpenter, L. J.: Quantifying the contribution of marine organic gases to atmospheric iodine, Geophys. Res. Lett., 37, L18804, https://doi.org/10.1029/2010GL043990, 2010.
Kerrigan, E. A., Kienast, M., Thomas, H., and Wallace, D. W. R.: Using oxygen isotopes to establish freshwater sources in Bedford Basin, Nova Scotia, a Northwestern Atlantic fjord, Estuarine, Coast. Shelf Sci., 199, 96–104, 2017.
Klick, S.: Seasonal variations of biogenic and anthropogenic halocarbons in seawater from a coastal site, Limnol. Oceanogr., 37, 1579–1585, 1992.
Kurihara, M. K., Kimura, M., Iwamoto, Y., Narita, Y., Ooki, A., Eum, Y. J., Tsuda, A., Suzuki, K., Tani, Y., Yokouchi, Y., Uematsu, M., and Hashimoto, S.: Distributions of short-lived iodocarbons and biogenic trace gases in the open ocean and atmosphere in the western North Pacific, Mar. Chem., 118, 156–170, https://doi.org/10.1016/j.marchem.2009.12.001, 2010.
Li, B.: Changes in Planktonic Microbiota, in Preserving the enviroment of Halifax Harbour, edited by: Willianm, L. K. W., Turner, G., and Ducharme, A., Fisheries and Oceans Canada, Halifax, 105–121, 2001.
Li, W. K. W.: Annual average abundance of heterotrophic bacteria and Synechococcus in surface ocean waters, Limnol. Oceanogr., 43, 1746–1753, 1998.
Liss, P. S. and Slater, P. G.: Flux of Gases across the Air-Sea Interface, Nature, 247, 181–184, https://doi.org/10.1038/247181a0, 1974.
Mahajan, A. S., Plane, J. M. C., Oetjen, H., Mendes, L., Saunders, R. W., Saiz-Lopez, A., Jones, C. E., Carpenter, L. J., and McFiggans, G. B.: Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic Ocean, Atmos. Chem. Phys., 10, 4611–4624, https://doi.org/10.5194/acp-10-4611-2010, 2010.
Mahajan, A. S., Gómez Martín, J. C., Hay, T. D., Royer, S.-J., Yvon-Lewis, S., Liu, Y., Hu, L., Prados-Roman, C., Ordóñez, C., Plane, J. M. C., and Saiz-Lopez, A.: Latitudinal distribution of reactive iodine in the Eastern Pacific and its link to open ocean sources, Atmos. Chem. Phys., 12, 11609–11617, https://doi.org/10.5194/acp-12-11609-2012, 2012.
Manley, S. L. and de la Cuesta, J. L.: Methyl iodide production from marine phytoplankton cultures, Limnol. Oceanogr., 42, 142–147, 1997.
Martino, M., Liss, P. S., and Plane, J. M. C.: The Photolysis of Dihalomethanes in Surface Seawater, Environ. Sci. Technol., 39, 7097–7101, https://doi.org/10.1021/es048718s, 2005.
Martino, M., Liss, P. S., and Plane, J.: Wavelength-dependence of the photolysis of diiodomethane in seawater, Geophys. Res. Lett., 33, L06606, https://doi.org/10.1029/2005GL025424, 2006.
Martino, M., Mills, G. P., Woeltjen, J., and Liss, P. S.: A new source of volatile organoiodine compounds in surface seawater, Geophys. Res. Lett., 36, L01609, https://doi.org/10.1029/2008GL036334, 2009.
McFiggans, G., Plane, J., Allan, B. J., Carpenter, L. J., Coe, H., and O'Dowd, C.: A modeling study of iodine chemistry in the marine boundary layer, J. Geophys. Res.-Atmos., 105, 14371–14385, 2000.
McFiggans, G., Coe, H., Burgess, R., Allan, J., Cubison, M., Alfarra, M. R., Saunders, R., Saiz-Lopez, A., Plane, J. M. C., Wevill, D., Carpenter, L., Rickard, A. R., and Monks, P. S.: Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701–713, https://doi.org/10.5194/acp-4-701-2004, 2004.
Moore, R. M. and Tokarczyk, R.: Volatile biogenic halocarbons in the northwest Atlantic, Global Biogeochem. Cy., 7, 195–210, 1993.
Moore, R. M. and Zafiriou, O. C.: Photochemical production of methyl iodide in seawater, J. Geophys. Res.-Atmos., 99, 16415–16420, 1994.
Moore, R. M., Geen, C. E., and Tait, V. K.: Determination of Henry's law constants for a suite of naturally occurring halogenated methanes in seawater, Chemosphere, 30, 1183–1191, 1995.
Mössinger, J. C., Shallcross, D. E., and Cox, R. A.: UV–VIS absorption cross-sections and atmospheric lifetimes of CH2Br2, CH2I2 and CH2BrI, J. Chem. Soc. Faraday Trans., 94, 1391–1396, 1998.
Nightingale, P. D., Malin, G., Law, C. S., Watson, A. J., Liss, P. S., Liddicoat, M. I., Boutin, J., and Upstill-Goddard, R. C.: In situ evaluation of air–sea gas exchange parameterizations using novel conservative and volatile tracers, Global Biogeochem. Cy., 14, 373–387, 2000.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hameri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Orlikowska, A. and Schulz-Bull, D. E.: Seasonal variations of volatile organic compounds in the coastal Baltic Sea, Environ. Chem., 6, 495–507, https://doi.org/10.1071/EN09107, 2009.
Orlikowska, A., Stolle, C., Pollehne, F., Jürgens, K., and Schulz-Bull, D. E.: Dynamics of halocarbons in coastal surface waters during short term mesocosm experiments, Environ. Chem., 12, 515–525, https://doi.org/10.1071/EN14204, 2015.
Rasmussen, R. A., Khalil, M. A. K., Gunawardena, R., and Hoyt, S. D.: Atmospheric methyl iodide (CH3I), J. Geophys. Res.-Oceans, 87, 3086–3090, 1982.
Rattigan, O. V., Shallcross, D. E., and Cox, R. A.: UV absorption cross-sections and atmospheric photolysis rates of CF3I, CH3I, C2H5I and CH2ICl, J. Chem. Soc. Faraday Trans., 93, 2839–2846, 1997.
Richter, U. and Wallace, D. W. R.: Production of methyl iodide in the tropical Atlantic Ocean, Geophys. Res. Lett., 31, L23S03, https://doi.org/10.1029/2004GL020779, 2004.
Saiz-Lopez, A. and Von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448, https://doi.org/10.1039/c2cs35208g, 2012.
Schall, C., Laturnus, F., and Heumann, K. G.: Biogenic volatile organoiodine and organobromine compounds released from polar macroalgae, Chemosphere, 28, 1315–1324, https://doi.org/10.1016/0045-6535(94)90076-0, 1994.
Shan, S., Sheng, J., Thompson, K. R., and Greenberg, D. A.: Simulating the three-dimensional circulation and hydrography of Halifax Harbour using a multi-nested coastal ocean circulation model, Ocean Dynam., 61, 951–976, 2011.
Shi, Q. and Wallace, D.: A three-year time-series of volatile organic iodocarbons in Bedford Basin, Nova Scotia: A Northwestern Atlantic Fjord, Mendeley Data, v1, http://dx.doi.org/10.17632/5y2t4mkjbv.1, 2018.
Shi, Q., Petrick, G., Quack, B., Marandino, C., and Wallace, D. W. R.: A time series of incubation experiments to examine the production and loss of CH3I in surface seawater, J. Geophys. Res.-Oceans, 2, 1022–1037, https://doi.org/10.1002/2013JC009415, 2014a.
Shi, Q., Petrick, G., Quack, B., Marandino, C., and Wallace, D.: Seasonal variability of methyl iodide in the Kiel Fjord, J. Geophys. Res.-Oceans, 119, 1609–1620, https://doi.org/10.1002/2013JC009328, 2014b.
Shimizu, Y., Ooki, A., Onishi, H., Takatsu, T., Tanaka, S., Inagaki, Y., Suzuki, K., Kobayashi, N., Kamei, Y., and Kuma, K.: Seasonal variation of volatile organic iodine compounds in the water column of Funka Bay, Hokkaido, Japan, J. Atmos. Chem., 74, 205–225, 2017.
Smythe-Wright, D., Boswell, S. M., Breithaupt, P., Davidson, R. D., Dimmer, C. H., and Eiras Diaz, L. B.: Methyl iodide production in the ocean: Implications for climate change, Global Biogeochem. Cy., 20, GB3003, https://doi.org/10.1029/2005GB002642, 2006.
Solomon, S., Garcia, R. R., and Ravishankara, A. R.: On the role of iodine in ozone depletion, J. Geophys. Res.-Atmos., 99, 20491–20499, 1994.
Stemmler, I., Rothe, M., Hense, I., and Hepach, H.: Numerical modelling of methyl iodide in the eastern tropical Atlantic, Biogeosciences, 10, 4211–4225, https://doi.org/10.5194/bg-10-4211-2013, 2013.
Stemmler, I., Hense, I., Quack, B., and Maier-Reimer, E.: Methyl iodide production in the open ocean, Biogeosciences, 11, 4459–4476, https://doi.org/10.5194/bg-11-4459-2014, 2014.
Tegtmeier, S., Krüger, K., Quack, B., Atlas, E., Blake, D. R., Boenisch, H., Engel, A., Hepach, H., Hossaini, R., Navarro, M. A., Raimund, S., Sala, S., Shi, Q., and Ziska, F.: The contribution of oceanic methyl iodide to stratospheric iodine, Atmos. Chem. Phys., 13, 11869–11886, https://doi.org/10.5194/acp-13-11869-2013, 2013.
Yamamoto, H., Yokouchi, Y., Otsuki, A., and Itoh, H.: Depth profiles of volatile halogenated hydrocarbons in seawater in the Bay of Bengal, Chemosphere, 45, 371–377, https://doi.org/10.1016/S0045-6535(00)00541-5, 2001.
Yokouchi, Y., Osada, K., Wada, M., Hasebe, F., Agama, M., Murakami, R., Mukai, H., Nojiri, Y., Inuzuka, Y., and Toom-Sauntry, D.: Global distribution and seasonal concentration change of methyl iodide in the atmosphere, J. Geophys. Res.-Atmos., 113, D18311, https://doi.org/10.1029/2008JD009861, 2008.
Yokouchi, Y., Saito, T., Ooki, A., and Mukai, H.: Diurnal and seasonal variations of iodocarbons (CH2ClI, CH2I2, CH3I, and C2H5I) in the marine atmosphere, J. Geophys. Res.-Atmos., 116, D06301, https://doi.org/10.1029/2010JD015252, 2011.
Ziska, F., Quack, B., Abrahamsson, K., Archer, S. D., Atlas, E., Bell, T., Butler, J. H., Carpenter, L. J., Jones, C. E., and Harris, N. R. P.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 13, 8915–8934, https://doi.org/10.5194/acp-13-8915-2013, 2013.